
神经发育障碍伴痉挛性双侧瘫痪和视觉缺陷(neurodevelopmental disorder with spastic diplegia and visual defects,NEDSDV;OMIM#615075)是一种非常罕见的常染色体显性遗传病,主要临床表现为严重的智力障碍、神经语言障碍、痉挛性双侧瘫痪、斜视、肌张力异常、自闭症行为、婴儿期发病等一系列神经系统异常[1-2]。CTNNB1基因是引起NEDSDV的一个关键的候选基因,位于3号染色体,包含14个外显子,编码包含781个氨基酸残基的β1连环蛋白(catenin beta1),该蛋白是钙黏蛋白复合物的一个组成部分,介导细胞间黏附,也是典型Wnt信号通路的关键下游分子。Wnt作为信号分子,在大脑发育中起着重要的作用。本文通过全外显子组测序(whole exome sequencing,WES)技术和Sanger测序对1个家系进行致病性基因变异的检测分析,明确其可能的致病原因。
1 病例报告
先证者 男,5岁,因严重的智力障碍、语言障碍、痉挛性双侧瘫痪、斜视、特殊面容,于2020年5月就诊于钦州市妇幼保健院(我院)。先证者母亲症状与先证者相似,其父母非近亲结婚,先证者为其母亲的第一胎,家属代诉其妊娠期无异常,为足月剖宫产。先证者外祖父母表型未见明显异常,否认类似家族遗传病史。查体:先证者上唇唇红较薄、长人中、鼻梁低平、鼻子突兀,双眼内斜(内聚),多斜视;可说长约3~4个字的句子,清音时模糊不清;屈髋,膝反张,双下肢肌张力增高,可独行,步态不稳,上下楼梯需扶扶手;手抖,抓握欠佳,语言理解、表达差,反应欠佳,无刻板怪异行为,可自动娱乐,碰到不会的东西易哭泣,可哄。家属代诉,先证者母亲症状与先证者相似,可听懂简单的指令,行走较先证者平稳,情绪较先证者稳定。先证者视力检查示屈光异常,眼诱发电位示双眼振幅下降,眼底检查未见异常。脑电图检查示小儿睡眠脑电图中度异常,脑地形图中度异常。既往检查血常规、血糖、肝肾功能、电解质等均未见异常。格塞尔发育量表(Gesell developmental schedule)提示重度发育迟缓,发育商分别为适应性32、大运动35、精细运动30、语言41、个人-社交35。本研究通过我院医学伦理委员会审查[编号:QSFY(2024)07.2],监护人签署了临床研究知情同意书。
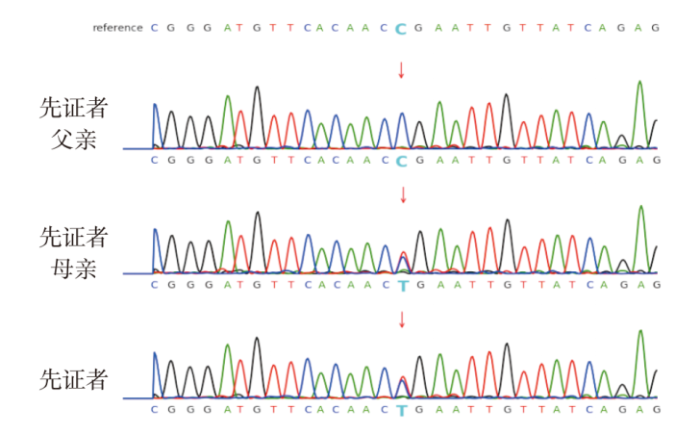
2021年5月该家系成员行WES,结果提示先证者及其母亲CTNNB1基因存在1个杂合截短变异c.1759C>T(p.R587*),此序列变化导致CTNNB1基因第1759位核苷酸的胞嘧啶(C)突变为胸腺嘧啶(T),导致第587位氨基酸变为终止密码子。先证者外祖父母及先证者父亲、羊水的检测结果未检测到变异。先证者及其父母亲的WES结果与Sanger测序结果是一致的(见图1),先证者所携带的CTNNB1基因杂合变异考虑为遗传自与其症状相似的母亲。而先证者的外祖父母未检测到该变异,考虑先证者母亲为新发变异。
图1
SWISS-MODEL软件在线预测分析野生型与p.R587*变异型蛋白质模型。模型显示CTNNB1基因c.1759C>T变异位点导致蛋白质结构发生改变,产生截短蛋白(见图2),明显影响蛋白活性。
图2
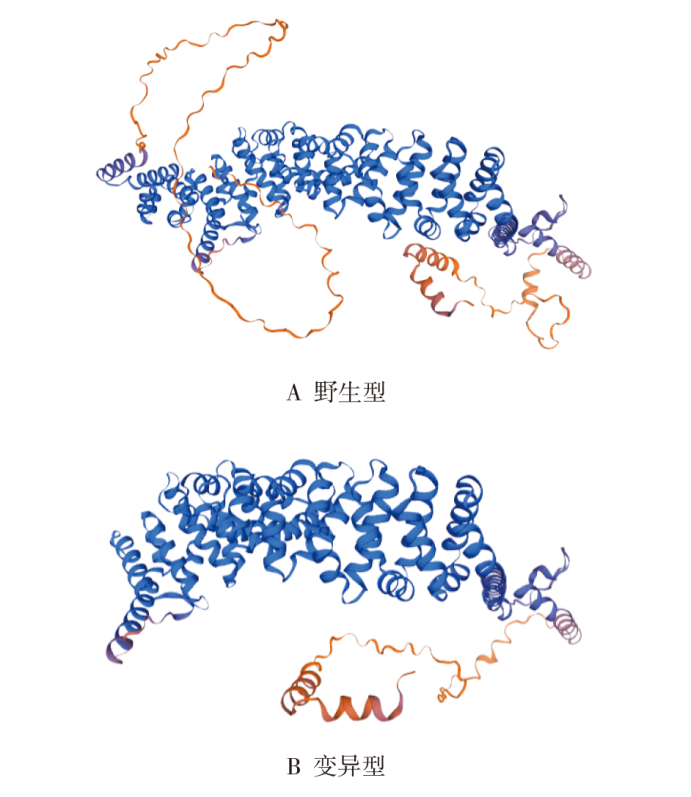
图2
SWISS-MODEL模拟CTNNB1基因编码蛋白序列的三维结构图
注:CTNNB1基因c.1759C>T变异后,产生截短蛋白,氨基酸R587后的氨基酸未进行翻译。
根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)《遗传变异分类标准与指南》,CTNNB1基因c.1759C>T变异判读为致病性变异(PVS1+PS2+PM2-Supporting),具体证据项如下:先证者及其母亲携带的CTNNB1基因c.1759C>T(p.R587*)杂合变异为截短变异,可导致第587位氨基酸变为终止密码子,产生一个截短蛋白,导致蛋白质序列丢失,从而导致蛋白功能丧失(PVS1);该基因位点变异可导致智力障碍、言语障碍及视力障碍等(PS2);该变异在基因组突变频率数据库、外显子组整合数据库和千人基因组数据库的正常对照人群中未见记录(PM2-Supporting)。
后续患儿不定期住院治疗,给予营养脑细胞、康复训练等治疗,发育无明显进步。截至2024年6月,随访患儿仍存在严重的智力障碍、语言障碍、痉挛性双侧瘫痪、斜视等,生活不能自理。
2 讨论
CTNNB1基因位于3号染色体p22.1区带,包含14个外显子,全长23.2 kb。CTNNB1编码的是一种高度保守的多功能连环蛋白,通过与细胞、黏附蛋白、信号分子和转录因子相互作用实现关键的细胞功能[10]。β1连环蛋白不仅是钙黏蛋白复合物的核心成分,也是典型Wnt信号传导的关键因子,在干细胞更新以及胚胎发生过程中的细胞增殖和分化中发挥重要作用[11]。本例先证者CTNNB1基因变异为c.1759C>T(p.R587*),为遗传自有症状的母亲。CTNNB1基因该位点的变异已在多篇文献中均有报道[1-
本研究中这对母子均具有NEDSDV的关键表型,如发育迟缓、智力残疾和视觉缺陷等。先证者格塞尔发育量表表示重度发育迟缓,5岁龄仍行走不稳,语言理解、表达差,提示该病的预后较差。目前在CTNNB1相关的神经发育障碍中尚未发现基因型-表型相关性[5],其中大多为散发不相关的病例。值得注意的是,本研究先证者遗传自有症状的母亲,该母子的临床表型的严重程度基本一致,这可能与遗传背景、环境因素等有一定的关系。该母子存在c.1759C>T(p.R587*)的变异,不排除这个氨基酸位置可能是CTNNB1的脆弱位点。本例患儿出现异常的脑电图,且该患儿无窒息抢救史,排除缺氧缺血性脑病和感染性疾病后,考虑为遗传性疾病所致。但在文献报道中极少发现有异常脑电图的病例[3-
此外,NEDSDV的诊断主要通过分子遗传学检测,WES检测可提高诊断率。NEDSDV无特异性治疗,多对症治疗及个体化综合康复治疗,包括运动功能、认知功能的训练和理疗等,尽可能减轻发育落后的程度,但本例患儿经过相应治疗后,效果不理想。因此,对于先证者父母如需再生育,应进行遗传咨询和产前诊断,避免再次生育该病的患儿。
综上,本研究报道了1对母子CTNNB1基因的截短变异c.1759C>T(p.R587*),该变异导致其出现了严重的智力障碍、语言障碍、痉挛性双侧瘫痪和斜视等。因此,CTNNB1变异与智力残疾、语言障碍等孤独症谱系障碍相关,有必要进行全面系统的遗传学检查,以确定致病的基因,为今后的遗传咨询及产前诊断提供依据。
参考文献
Clinical features associated with CTNNB1 de novo loss of function mutations in ten individuals
[J].
Loss of function mutations in CTNNB1 have been reported in individuals with intellectual disability [MIM #615075] associated with peripheral spasticity, microcephaly and central hypotonia, suggesting a recognisable phenotype associated with haploinsufficiency for this gene. Trio based whole exome sequencing via the Deciphering Developmental Disorders (DDD) study has identified eleven further individuals with de novo loss of function mutations in CTNNB1. Here we report detailed phenotypic information on ten of these. We confirm the features that have been previously described and further delineate the skin and hair findings, including fair skin and fair and sparse hair with unusual patterning.Copyright © 2016 Elsevier Masson SAS. All rights reserved.
CTNNB1基因变异致伴有痉挛性双瘫和视觉缺陷的神经发育障碍5例报告并文献复习
[J].
Somatic mutations of GNA11 and GNAQ in CTNNB1-mutant aldosterone-producing adenomas presenting in puberty, pregnancy or menopause
[J].Most aldosterone-producing adenomas (APAs) have gain-of-function somatic mutations of ion channels or transporters. However, their frequency in aldosterone-producing cell clusters of normal adrenal gland suggests a requirement for codriver mutations in APAs. Here we identified gain-of-function mutations in both CTNNB1 and GNA11 by whole-exome sequencing of 3/41 APAs. Further sequencing of known CTNNB1-mutant APAs led to a total of 16 of 27 (59%) with a somatic p.Gln209His, p.Gln209Pro or p.Gln209Leu mutation of GNA11 or GNAQ. Solitary GNA11 mutations were found in hyperplastic zona glomerulosa adjacent to double-mutant APAs. Nine of ten patients in our UK/Irish cohort presented in puberty, pregnancy or menopause. Among multiple transcripts upregulated more than tenfold in double-mutant APAs was LHCGR, the receptor for luteinizing or pregnancy hormone (human chorionic gonadotropin). Transfections of adrenocortical cells demonstrated additive effects of GNA11 and CTNNB1 mutations on aldosterone secretion and expression of genes upregulated in double-mutant APAs. In adrenal cortex, GNA11/Q mutations appear clinically silent without a codriver mutation of CTNNB1.© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc.
Case Report: A de novo CTNNB1 Nonsense Mutation Associated With Neurodevelopmental Disorder, Retinal Detachment, Polydactyly
[J].
Missense variants in CTNNB1 can be associated with vitreoretinopathy-Seven new cases of CTNNB1-associated neurodevelopmental disorder including a previously unreported retinal phenotype
[J].
Diagnostic exome sequencing in persons with severe intellectual disability
[J].
De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum
[J].Recently, de novo heterozygous loss-of-function mutations in beta-catenin (CTNNB1) were described for the first time in four individuals with intellectual disability (ID), microcephaly, limited speech and (progressive) spasticity, and functional consequences of CTNNB1 deficiency were characterized in a mouse model. Beta-catenin is a key downstream component of the canonical Wnt signaling pathway. Somatic gain-of-function mutations have already been found in various tumor types, whereas germline loss-of-function mutations in animal models have been shown to influence neuronal development and maturation. We report on 16 additional individuals from 15 families in whom we newly identified de novo loss-of-function CTNNB1 mutations (six nonsense, five frameshift, one missense, two splice mutation, and one whole gene deletion). All patients have ID, motor delay and speech impairment (both mostly severe) and abnormal muscle tone (truncal hypotonia and distal hypertonia/spasticity). The craniofacial phenotype comprised microcephaly (typically -2 to -4 SD) in 12 of 16 and some overlapping facial features in all individuals (broad nasal tip, small alae nasi, long and/or flat philtrum, thin upper lip vermillion). With this detailed phenotypic characterization of 16 additional individuals, we expand and further establish the clinical and mutational spectrum of inactivating CTNNB1 mutations and thereby clinically delineate this new CTNNB1 haploinsufficiency syndrome.
Genomic and phenotypic characterization of 404 individuals with neurodevelopmental disorders caused by CTNNB1 variants
[J].Germline loss-of-function variants in CTNNB1 cause neurodevelopmental disorder with spastic diplegia and visual defects (NEDSDV; OMIM 615075) and are the most frequent, recurrent monogenic cause of cerebral palsy (CP). We investigated the range of clinical phenotypes owing to disruptions of CTNNB1 to determine the association between NEDSDV and CP.Genetic information from 404 individuals with collectively 392 pathogenic CTNNB1 variants were ascertained for the study. From these, detailed phenotypes for 52 previously unpublished individuals were collected and combined with 68 previously published individuals with comparable clinical information. The functional effects of selected CTNNB1 missense variants were assessed using TOPFlash assay.The phenotypes associated with pathogenic CTNNB1 variants were similar. A diagnosis of CP was not significantly associated with any set of traits that defined a specific phenotypic subgroup, indicating that CP is not additional to NEDSDV. Two CTNNB1 missense variants were dominant negative regulators of WNT signaling, highlighting the utility of the TOPFlash assay to functionally assess variants.NEDSDV is a clinically homogeneous disorder irrespective of initial clinical diagnoses, including CP, or entry points for genetic testing.Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
CTNNB1基因突变致智力障碍3例患儿临床特点及文献复习
[J].
Wnt signaling from membrane to nucleus: β-catenin caught in a loop
[J].β-catenin is the central nuclear effector of the Wnt signaling pathway, and regulates other cellular processes including cell adhesion. Wnt stimulation of cells culminates in the nuclear translocation of β-catenin and transcriptional activation of target genes that function during both normal and malignant development. Constitutive activation of the Wnt pathway leads to inappropriate nuclear accumulation of β-catenin and gene transactivation, an important step in cancer progression. This has generated interest in the mechanisms regulating β-catenin nuclear accumulation and retention. Here we discuss recent advances in understanding feedback loops that trap β-catenin in the nucleus and provide potential insights into Wnt signaling and the development of anti-cancer drugs.Copyright © 2012 Elsevier Ltd. All rights reserved.
Wnt signaling in development and tissue homeostasis
[J].
Dominant β-catenin mutations cause intellectual disability with recognizable syndromic features
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}